
MDR過渡條款(MDD展延)-實務經驗分享
2023 年3月20日歐盟執委會公布的2023/607,延長了MDR過渡時間軸,讓製造商在申請轉證MDR的過程中繼續維持醫材在歐洲市場流通。
但並非所有的MDD產品皆適用,其中包含相當多的限制;Luanamed透過實際與主管機關交涉案例,與大家分享申請的步驟與細節,替業界先進描繪更完整的MDR過渡 (MDD展延)全貌:
1. 製造廠得依據2023/607 進行MDR過渡(MDD展延)的適用條件
首先來了解,誰適用過渡條款?
只有在2021年5月21日前,根據90/385/EEC(AIMD)和93/42/EEC(MDD)所頒發的證書或符合性聲明(DoC)所涵蓋的Legacy Device產品,並且提出具體MDR轉證計劃,才適用過渡條款範圍。
除此,MDD證書到期日必須落在2021年5月26日後,才有機會進行MDD展延。詳細的關鍵日期判定,您可再參考這篇:《5分鐘搞清楚MDR過渡條款(MDD展延),到底誰適用?》。
依據該文章,MDD展延有三條路徑,本文將討論:MDD證書在2023年3月20日前到期,並且在證書到期日前未與NB簽約轉版MDR時,必須向成員國主管機關申請符合性豁免的路徑。
2. 過渡條款之法之依據
在MDR article 59,說明針對符合性途徑的豁免(Derogation from the conformity assessment procedures),包含:
- 透過Article 59,歐盟境內主管機關(Competent Authority,後續稱CA)可以在正當理由、且有利於公共健康與病患安全前提下,無須等待符合性途徑(Article 52)完備,先於成員國境內將醫療器材上市。
- 如果該醫療器材非僅用於單一病患,該成員國必須將此上市決議,通知歐盟執委會以其他成員國,執委會將針對病患的安全或健康狀況,一定程度地展延成員國在歐盟境內授權的時限,以及設置該醫療器材使用之條件。
MDR article 59有前提的放寛了尚未完成MDR程序的醫療器材,而MDR Article 120過渡條款,更提出透過MDD或AMID路徑取得的證書,在未有重大的設計變更下,製造商可於寬限期內,持續將醫療器材/體外診斷試劑使用或投放於歐盟市場,直至2024年5月26日為止。
這段期間製造商必須進行MDR轉版,並著手向CA提出展延申請,才能使該MDD證書延長至2027或2028年(依分類分級而異)。
3. 展延流程實例分享(以荷蘭CA為例)
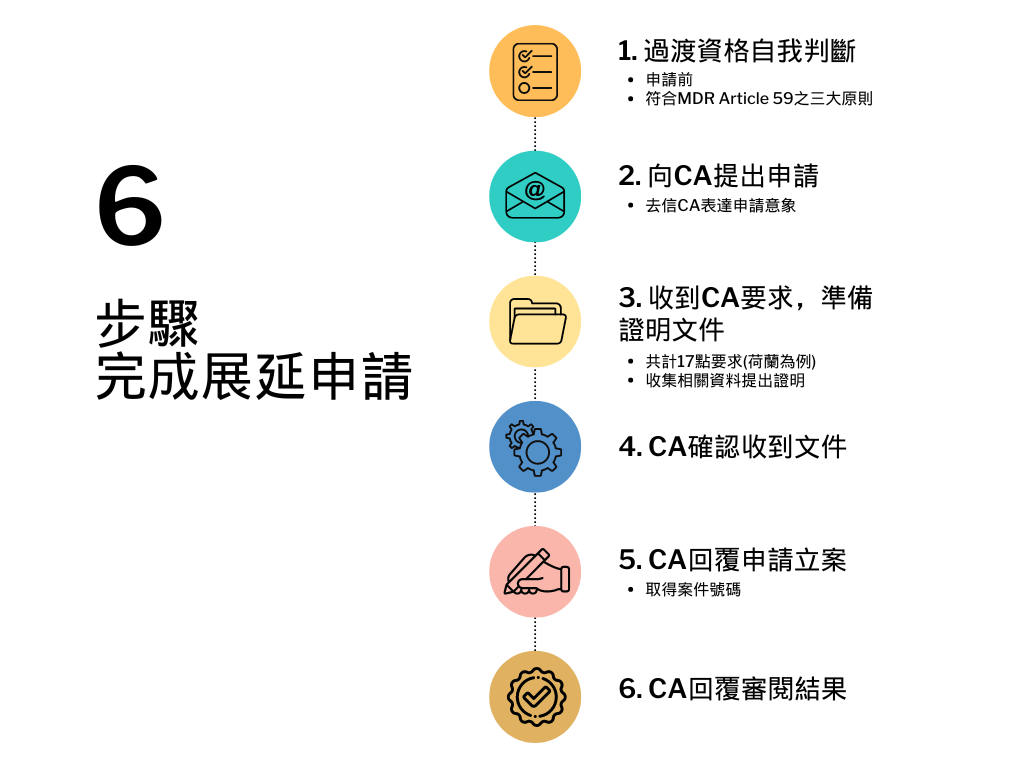
當製造廠有計畫的規劃轉證,且原有MDD證書到期日也允許進行展延,即可向主管機關提出申請,以下為實際與荷蘭CA申請流程:
- 製造商自行判斷過渡期資格及路徑:
- 時間點:向CA申請前。
- 檢視產品是否包含下列三大原則:
- 「若未予以上市,將對病患照護帶來不可接受的風險」--病患福祉,符合Article 59--Without a derogation, an unacceptable risk will arise for patient care.
- 「沒有可用的等效替代產品」--病患福祉,符合Article 59--No equivalent alternative is available.
- 「製造商已經合理地表明該產品為安全,且醫療器材之品質以及安全性皆能符合相關法規要求」--需證實該醫材/體外診斷試劑除符合MDD外,亦有具體MDR轉證計劃,符合Article原始精神--The manufacturer has made it plausible that the device is safe and that the MD/IVD meets the quality and safety requirements as described in the relevant legislation and regulations.
- 向CA提出申請:
- 簡易去信CA表達申請意象:希望遵循MDR Article 59, first paragraph 1或IVDR Article 54, first paragraph來申請豁免。
- CA回覆要求,依據要求提出文件說明<細節見下一段落”文件要求”>
- CA收件,確認收到文件
- CA回覆申請立案,取得案件號碼
- CA回覆審閱結果
4. 文件要求說明
由CA提出的17點要求,申請者須逐一說明並提供相關文件(包含文件名稱與編號),藉此作為三大原則之證據:
A. 「若未予以上市,將對病患照護帶來不可接受的風險」Unacceptable Risk for patient care
B. 「沒有可用的等效替代產品」No alternative
C. 「該醫材的品質以及安全性皆能符合相關法規要求」Quality/Safety meet regulation
| 項次 | 要求 | A | B | C |
|---|---|---|---|---|
| 1 | 若申請者已在EUDAMED註冊,請提供申請者的SRN。 If applicable: when you, as the applicant are already registered in EUDAMED your SRN. 請包含:申請商的基本資料(例如國家、機能)、製造商的基本資料,並確認是否計劃於歐盟上市的醫療器材/體外診斷試劑(Manufacturer intends to CE mark the MD / IVD)。 另外,也請提供最終的展延決議接收人資訊(名稱、電子郵件、地址等)。 | ○ | ||
| 2 | 概述申請於[荷蘭]市場銷售的產品。 An overview of the products for which you, as the applicant, apply for derogation and which have already been placed on the market in the Netherlands. 請包含:產品名稱、型號與產品預期目的(Intended purpose of the MD/IVD),以及DoC等。 | ○ | ○ | ○ |
| 3 | 說明原始CE證書無法延長,或需要過渡的原因。 The reason that you as, an applicant, need a derogation and/or why the CE certificate could not be extended in time. | ○ | ○ | |
| 4 | 概述是否已向所在國家CA提出類似的展延申請,以及CA給予的決定與理由。 An overview of the competent authorities within the EU from whom you, as the applicant, have applied for a similar derogation, including the decision and reasoning of the relevant competent authorities. | ○ | ||
| 5 | NB預計發放CE證書的日期,包括NB已確定的時程。 The expected date that the CE certificate will be issued by the Notified Body, including the timeframe confirmed by the Notified Body. | ○ | ||
| 6 | 提供預計申請展延的醫療器材/體外診斷試劑,在過去2年內在[荷蘭]銷售概況。包括: A sales overview of the MD/IVD’S that fall under the application for derogation and that have been sold in the Netherlands in the past 2 years. Among which: -各產品類型的數量,包括客戶及護理照護者; - the numbers per type of product, including customers/care providers, and; -與使用數據結合的庫存狀況,得到關鍵照護者庫存天數。 - the stock situation in combination with usage figures resulting in the number of days of stock at the most critical care providers. | ○ | ○ | |
| 7 | 如果您的CE證書已經過期,請提供您通知客戶/醫療機構該證書過期的證據。 If applicable: if your CE certificate has already expired, proof that customers / healthcare institutions have been informed about the invalidity of the CE certificate. | ○ | ||
| 8 | 提供至少由一家醫療機構使用醫療器材/體外診斷試劑的基本重要性聲明書/宣告書。 A statement/ declaration of the essential importance of the use of the MD/IVD by at least 1 health institution. | ○ | ○ | |
| 9 | 說明如果本展延未能獲得核准,對於病患安全性的風險影響。 An explanation of what you, as the applicant, believe the risks are with regard to patient safety if the requested derogation is not granted. | ○ | ○ | |
| 10 | 提供在荷蘭受影響病患人口數的估算。 An estimate of the affected patient population in the Netherlands. | ○ | ○ | |
| 11 | 勾選「如果未獲得展延,對病患產生的效果」: Tick the corresponding box to what the effect will be on the patient if no derogation is granted. ☐ 輕微傷害或暫時性生活品質的輕微降低 ☐ Minor injury or minor reduction in quality of life of a temporary nature ☐ 有限傷害或不可逆的生活品質有限降低 ☐ Limited injury or limited reduction in quality of life of an irreversible nature ☐ 嚴重傷害或暫時性生活品質的嚴重降低 ☐ Serious injury or serious temporary loss of quality of life of a temporary nature ☐ 嚴重傷害或不可逆的生活品質嚴重降低 ☐ Serious injury or serious impairment of quality of life of an irreversible nature ☐ 生命威脅的傷害或無法接受的生活品質降低 ☐ Life-threatening injury or unacceptable reduction in quality of life ☐ 其他: ☐ Other: | ○ | ○ | |
| 12 | 勾選「如果未獲得展延,對病患影響的持續時間」: Tick the corresponding box regarding the duration of the impact on the patient if no derogation is granted: ☐ 1 天 ☐ 1 週 ☐ 1 月 ☐ 超過 1 月 ☐ 永久 ☐ 1 day ☐ 1 week ☐ 1 month ☐ more than a month ☐ permanen | ○ | ○ | |
| 13 | 概述如果未獲得展延,對荷蘭客戶(經銷商和/或醫療提供者)預期發生的風險,及發生的時間。 An overview of the Dutch customers (resellers and/or healthcare providers) with whom you, as the applicant, expect that these risks will occur if the requested derogation is not granted, including the timeline in which these risks are expected. | ○ | ○ | |
| 14 | 說明是否可以在短期內(由其他醫療器材製造商)替換所申請展延的醫療器材/體外診斷試劑,且不增加額外風險,並附上說明。 An explanation of whether the MD/IVD for which a derogation is requested can be replaced without additional risk in the short term by MD/IVD from other manufacturers, including a motivation. | ○ | ||
| 15 | 若適用:若NB對於申請展延產品中發現不符合事項,包含不符合事項被排除/解決的日期。 If applicable: An overview of the outstanding non-conformities that the Notified Body has found in the products for which you, as an applicant, apply for a derogation, including the expected end date when the non-conformities have been removed/ solved. | ○ | ||
| 16 | 提供相關CE證書的副本。 A copy of the relevant CE certificate. | ○ | ||
| 17 | 需確認此展延申請涵蓋的產品,在CE證書過期後未經過改變/修改,且在展延後仍以完全相同的條件生產及交付。 A confirmation that the products covered by this application for derogation have not been adapted/changed after the CE certificate has expired and will be produced and delivered under exactly the same conditions in the event of a possible derogation. | ○ |
申請前小提醒
強烈建議申請前,製造商必須先就「醫材轉證MDR的流程」與NB達成合意,最好雙方已完成簽約,或雙方合作意向書或書面溝通紀錄。
除了NB,也建議與歐盟境內代理商/經銷商合意,先行取得「醫療器材/體外診斷試劑的歐盟境內銷售紀錄」,其須包含醫療院所名稱、地址、器材/試劑使用數量等詳細訊息;以利後續回覆CA要求所需。
文件要求準備重點:
文件要求中,CA透過產品銷售資訊來判斷醫療器材對病患重要性;務必針對醫療器材/體外診斷試劑之銷售地點、醫院(診所名稱)、病患人數等資訊完整呈現,否則可能判定為不具展延資格。
建議製造商必須利用MDR法規要求的機會,要求經銷/代理商配合,提供基本銷售與使用者資訊列冊,以符合MDR法規。
獲得展延後,後續要注意甚麼?
過渡期僅到2024年5月26日,所以製造商務必在此之前完成MDR品質系統實踐,並提交NB審查申請。
NB需於2024/9/26前完成審查並確認合規,該MDD證書才可以延長到2027(Class IIb, Class III MD) or 2028(Class IIa, Class I MD)。



AUTHOR
&
CO-AUTHOR
Chen Jen-Ru
Y. H.